Resistance exercise: a mighty tool that adapts, destroys, rebuilds and modulates the molecular and structural environment of skeletal muscle
Article information
Abstract
[Purpose]
Skeletal muscle regulates health and performance by maintaining or increasing strength and muscle mass. Although the molecular mechanisms in response to resistance exercise (RE) significantly target the activation of protein synthesis, a plethora of other mechanisms and structures must be involved in orchestrating the communication, repair, and restoration of homeostasis after RE stimulation. In practice, RE can be modulated by variations in intensity, continuity and volume, which affect molecular responses and skeletal muscle adaptation. Knowledge of these aspects is important with respect to planning of training programs and assessing the impact of RE training on skeletal muscle.
[Methods]
In this narrative review, we introduce general aspects of skeletal muscle substructures that adapt in response to RE. We further highlighted the molecular mechanisms that control human skeletal muscle anabolism, degradation, repair and memory in response to acute and repeated RE and linked these aspects to major training variables.
[Results]
Although RE is a key stimulus for the activation of skeletal muscle anabolism, it also induces myofibrillar damage. Nevertheless, to increase muscle mass accompanied by a corresponding adaptation of the essential substructures of the sarcomeric environment, RE must be continuously repeated. This requires the permanent engagement of molecular mechanisms that re-establish skeletal muscle integrity after each RE–induced muscle damage.
[Conclusion]
Various molecular regulators coordinately control the adaptation of skeletal muscle after acute and repeated RE and expand their actions far beyond muscle growth. Variations of key resistance training variables likely affect these mechanisms without affecting muscle growth.
INTRODUCTION
Resistance exercise (RE) is the most effective training method to increase strength and muscle mass. Those factors are crucial for enhancing performance in elite sports, but also to maintain good health across the lifespan [1,2]. For instance, RE substantially improves insulin sensitivity, lowers the risk of type II diabetes [3], and increases skeletal muscle strength, thus reducing the risk of falling in older adults [4,5]. Thus, the World Health Organization (WHO) guidelines recommend the implementation of RE in everyone’s physical activity routines [6]. They propose conducting moderate intensity RE 2–3 times per week with weight loading for the major muscle groups. Considering the current data determining the specific aspects of RE–induced skeletal muscle adaptations with respect to intensity, volume and progression, this general recommendation is yet relatively vague. Nevertheless, it is highly valuable as it generally emphasizes the importance of RE and contributes to better acceptance of a training method that should be conducted in all age groups, including children [7,8].
Acute and chronic RE significantly affect skeletal muscle growth and strength [9], driving this adaptation depending on age, training volume and intensity [10,11]. However, RE also threatens the structural integrity of skeletal muscles [12,13]; therefore, proteostatic mechanisms are constantly engaged in order to maintain tissue integrity [14].
This narrative review introduces the major mechanisms of global and molecular muscle adaptation in response to RE that have been elucidated in numerous studies over the last decade. These mechanisms act on skeletal muscle to regulate its growth, structural remodeling and maintenance in response to acute and chronic RE. In particular, the molecular responses of skeletal muscle will be highlighted, that are rapidly activated after acute RE and that play a vital role for maintaining proteostasis, ensure the ability to structurally recover, and prime the muscle for repeated RE. We also addressed and implemented crucial aspects of training volume, intensity and continuity that modulate these mechanisms, influence the outcome, and inherit aspects that may be useful in estimating the training effects exerted by RE over extended time courses.
RE is characterized by muscle contractions against moderate-to-high resistance [9]. This requires the active recruitment of an extensive amount of muscle fibers, which is controlled by the central nervous system [15,16]. The recruited muscle fibers generate force through sarcomeric contractions, thereby exerting mechanical tension within the sarcomeric cytoskeleton [17]. Mechanical stimuli are accepted as key components of the complex stimulation pattern induced by RE and considered to induce protein synthesis and, in the long-term, muscle growth [18,19].
In molecular exercise physiology, considerable evidence has accumulated, underpinning the influence of external loads on muscle growth [20,21] and strength [22,23].
In order to increase strength in the aging and younger populations, RE should be performed with a relatively high force (>80% of the one-repetition maximum; 1RM), with approximately 8–12 repetitions [24,25]. However, in untrained individuals and older adults, a wider range of 6–15 repetitions, corresponding to 90–60% of the 1RM, can generate similar strength gains, at least for the first 6 months of RE training [26]. In contrast, RE that is carried out within a range of 40–60% of the 1RM has the potential to increase muscle hypertrophy similar to that observed at higher intensities of exercise [24,25].
In these studies, the number of repetitions with a lower training intensity was significantly higher than that observed with higher loading intensities, thereby increasing the time under tension. This implies that, despite the degree of tension (e.g., higher weight loading), an extended time under tension will also increase protein synthesis to an equal level [27,28]. This notion may also be valuable for the broad application of RE, when the idea of moving high weights until exhaustion causes anxiety and potentially discourages primarily a previously untrained population from participation in continuous RE programs or initiation in the first place.
Importantly, the induction of significant fatigue during RE with a higher volume or weight loading is assumed to be unnecessary [22,23,29]. In contrast, RE with lower weights (30% of 1RM) showed a greater effect on muscle hypertrophy when performed until failure versus not until failure [30]. Otherwise, to optimize muscle growth with moderate intensity, RE should at least be conducted until the movement speed of contractions is reduced [31]. Such a strategy ensures that muscle contractions approximate a point close to fatigue but do not actually reach it, thereby potentially reducing the time required for regeneration. However, a nonlinear relationship was assumed between proximity-to-failure and muscle hypertrophy [31].
The eccentric contraction phase during RE is characterized by the lengthening of the muscle, caused by external forces that are greater than the forces produced by the contracting and shortening muscle [32]. In particular, this eccentric contraction phase can induce muscle damage in the form of myofibrillar disruptions and lesions in the sarcomeric z-disk [33,34]. This is even more likely when the muscle is fatigued [35]. As a consequence of muscle damage, sensations of muscle soreness occur within the next few days; muscle force and performance are blunted, and the ability to continue further training sessions can be impaired [36].
However, repeated RE enables muscle cells to modulate the molecular muscle environment so that damage decreases gradually over time; this phenomenon is widely known as the “repeated bout effect” [37].
Mechanisms, such as chaperone-assisted selective autophagy (CASA) [38], immediate chaperone activation [39], and the unfolded protein response (UPR) [40], act within minutes in acutely damaged muscle and regulate in the long term the protection, refolding, degradation and expression of proteins that stabilize, e g., the sarcomeric z-disc [14]. Muscle repair and protein synthesis are time- and energy-consuming processes [41,42], during which growth-mediating pathways are transiently and locally disabled [43].
Muscle damage and the associated proteostatic mechanisms may, therefore, be responsible for the fact that substantial hypertrophy only follows after repeated stimulation of skeletal muscle, and after a time frame during which skeletal muscle has adapted so that extensive myofibrillar damage caused by RE is reduced [44].
General adaptations of skeletal muscle tissue induced by resistance exercise
RE has a significant effect on the composition of the entire skeletal muscle, expanding considerably beyond the dominantly occurring myofibrillar protein accretion that leads to hypertrophy. Although endurance training (ET) is the major stimulus that induces adaptations in oxidative metabolism, RE can acutely increase mitochondrial protein synthesis [27]. In addition, RE increased mitochondrial function in both trained and untrained younger subjects [45-47]. However, studies investigating the long-term increase in mitochondrial content in response to RE have shown equivocal results [47]. Consequently, RE is not considered to significantly enhance mitochondrial content. This might be because, compared to ET, RE does not primarily target the activation of signaling pathways controlling mitochondrial synthesis [48]. In contrast, damage-inducing exercise such as RE is more associated with the complex regulation of mitochondrial quality control via mitophagy [49].
Additionally, acute RE facilitates a rapid increase in sarcoplasmic protein synthesis [50] in skeletal muscle. This results in increased glycolytic enzyme abundance and anaerobic capacity [2,51], which is beneficial for supporting energy metabolism in power-associated sports [52]. However, although sarcoplasmic proteins may accumulate after repeated RE, only small proportions of sarcoplasmic proteins actually seem to change after continued strength training and the existence of sarcoplasmic hypertrophy is still discussed [21].
Another essential component of skeletal muscle is the extracellular matrix (ECM), which is a complex molecular network consisting of collagens, specialized cells, diverse glycoproteins and proteoglycans [53,54]. Within the skeletal muscle ECM, collagens form a network of intramuscular connective tissue (IMCT) that surrounds and connects muscle fibers (endomysium), fiber bundles (perimysium), and the entire skeletal muscle (epimysium) [55]. Collagen fibers are important components of the IMCT, and the dominant collagen isoform as well as the associated matrix composition differ considerably, depending on localization and function of the IMCT [54].
The endomysium is located between the basement membranes of two neighboring myofibers, where it senses and transduces particularly lateral contraction-induced forces [55,56]. Mechanical strain exerted by RE induces rapid collagen turnover in skeletal muscle [57]. Collagen degradation and synthesis are coordinately activated in skeletal muscle within 6 h after RE and synchronized with myofibrillar protein synthesis [58] to match long-term muscle growth, ECM and IMCT development. Whether IMCT increases after repeated RE training is still debated. Studies reported that it increased upon immobilization [59-61], and in older adults [62,63], whereas in younger people, IMCT content likely increases only in connection with muscle growth [55]. However, RE acutely stimulates and remodulates IMCT via mechanical strain to support angiogenesis [64], repair damaged IMCT [57], and reorganize its structure [65,66] when necessary.
The mechanical forces generated by the myofilaments (Figure 1 (1)) are transduced via lateral force transmission [67] at the z-discs and sarcomeric costameres to the ECM (Figure 1 (2)). The sarcomeric cytoskeleton is connected to the ECM by a complex network of force-transducing proteins17. Integrins, especially alpha7/beta1 [68], increased after continued RE and enhanced mTOR signaling and muscle growth in rats [69].
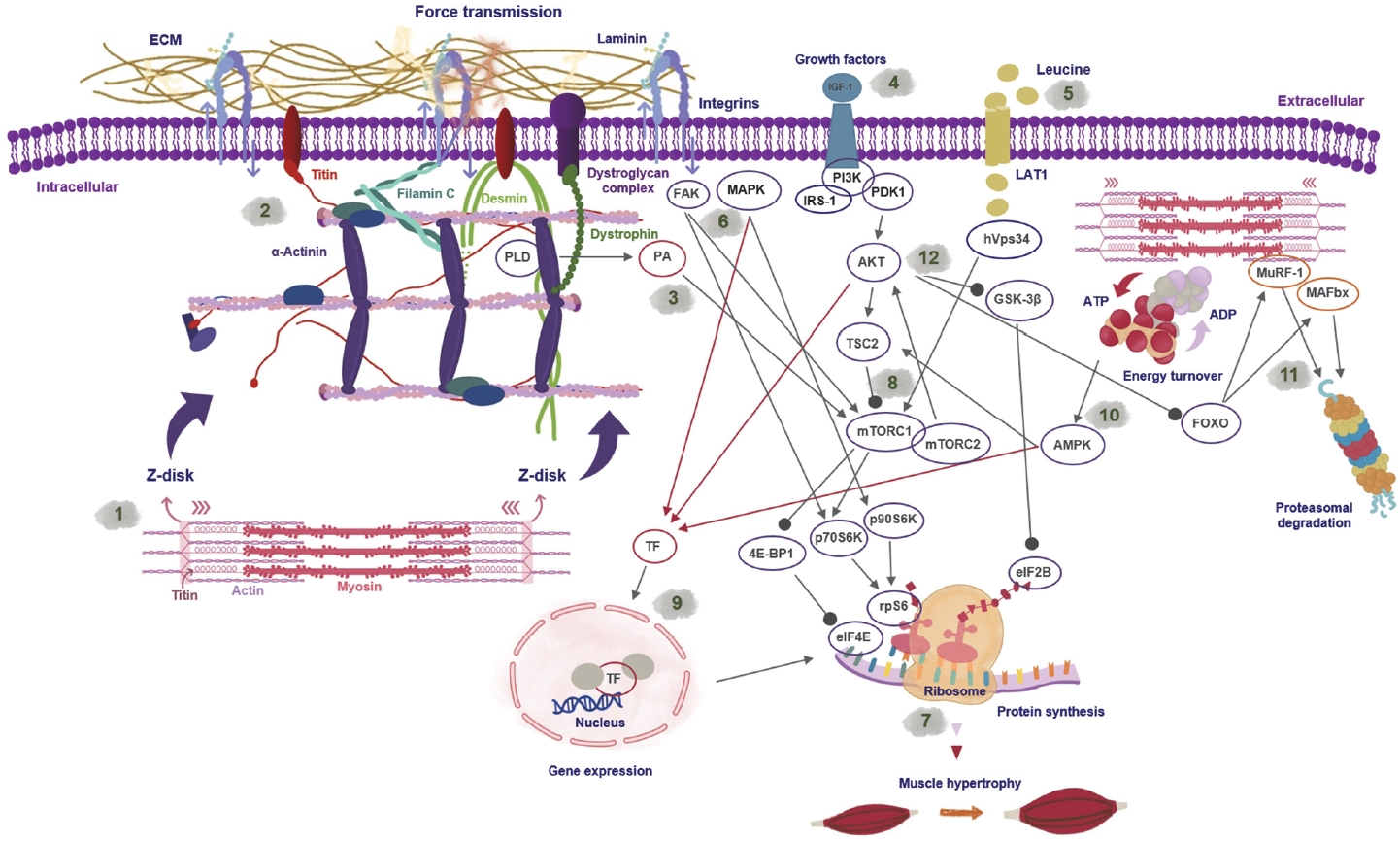
Schematic representation of the coupling between sarcomeric force development during contraction and the associated molecular signaling which controls gene expression, protein synthesis and degradation. (1) Sarcomeric contraction initiates force development that is laterally transduced via the sarcomeric cytoskeleton. Titin, a giant spring-like sarcomeric protein, senses mechanical forces and contributes to skeletal muscle adaptation by altering molecular signaling pathways. (2) Force transmission via integrins and the dystrophin-glycoprotein complex to the extracellular matrix (ECM). Mechanical stimulation of the sarcomeric environment activates mTOR (mammalian target of rapamycin)- signaling independently of AKT (protein kinase B). (3) Mechanical stimulation via contraction activates PLD (phospholipase D), generating phosphatidic acid, which activates mTOR signaling. (4) Growth factors like Insulin-like growth factor 1 (IGF-1) mediate the AKT-dependent activation of mTOR. (5) Leucine availability activates mTOR independent of growth factors. (6) Force-sensing integrins mediate focal adhesion kinase (FAK)-activation, which can activate p70S6K (70-kDa ribosomal protein S6 kinase) independent of mTOR. Additionally, MAP kinases activate p90S6K (90-kDa ribosomal protein S6 kinase), which phosphorylates rpS6 also independently of mTOR. (7) Protein synthesis (8) is predominantly activated by mTOR; however, other signaling components converge and control translation initiation. Activated mTOR (mTORC1/2 complex) controls protein synthesis by phosphorylating 4E-BP1 (4E-binding protein 1), relieving its inhibitory effect on eiF4E; mTOR further phosphorylates p70S6K, which phosphorylates and activates rpS6 (ribosomal protein S6). AKT activation phosphorylates and inhibits glycogen synthase kinase 3 (GSK3)-beta and relieves its inhibitory effect on eiF2B, which is required for translational regulation. (9) Molecular signaling kinases, e.g., mitogen-activated protein kinase (MAPK), AKT and adenosine monophosphate-activated protein kinase (AMPK), activate transcription factors, which enter the nucleus to initiate gene expression (red arrows). (10) Metabolic stress induced by increased adenosine triphosphate (ATP) turnover activates AMPK, which phosphorylates tuberous sclerosis complex 2 (TSC2) proteins that inhibit mTOR activity. (11) Forkhead-box (FOXO) proteins control the expression of muscle RING-finger 1 (MuRF-1) and muscle atrophy-F-box/ atrogin1 (MAFbx) proteins, and E3 ligases, which control the degradation of sarcomeric proteins via proteasomal degradation. (12) Growth factor signaling via insulin or IGF-1 phosphorylates and activates AKT, which inhibits FOXO proteins via phosphorylation and by that proteasomal degradation of sarcomeric proteins.
In a model of human RE, we determined that also z-disc proteins, such as filamin C (FLNC) and the intermediate filament desmin increase after 6 weeks of progressive RE (six sets of 8–12 repetitions for the knee extensors) in young subjects [12,70]. Both proteins contribute to the sarcomeric stability of the skeletal muscle z-disc [71,72] (Figure 1 (2)). The RE–induced increase in the expression of FLNC and desmin was associated with a decrease in RE–induced myofibrillar damage. Importantly, some ECM proteins also contribute to tissue remodeling after skeletal muscle damage. For instance, the mechanosensitive ECM protein tenascin-C was associated with increased muscle repair in a mouse model [73]. This highlighted the important role of the ECM of the skeletal muscle, which permanently interacts with skeletal muscle cells in response to mechanical stimulation. A recent publication excellently reviewed how dynamically the ECM adapts to diverse exercise modes [65].
The ECM also builds up the niche in which the muscle stem cells, “satellite cells” (SCs), are located between plasma and basement membrane around muscle fibers [74]. After exposure to specific signals, SCs attain the ability to proliferate and differentiate into myoblasts, which fuse with existing muscle fibers [74]. SCs also have the potential to maintain and repair muscles in older adults [75]. Mechanical strain on the SC niche, but also damage, growth factors and cytokines control the activation of SCs [76,77].
In the context of RE, moderate muscle damage appears to have beneficial effects on skeletal muscle adaptability and enhances satellite cell proliferation in skeletal muscle [78]. It has been shown that the SC pool expands after periods of RE training and supports the long-term growth of muscle [79,80]. SCs enhance the hypertrophic response of the human skeletal muscle to long-term RE by fusing with damaged myofiber areas and adding new myonuclei to the skeletal muscle [81]. Correspondingly, inhibition of SC proliferation blunts exercise-stimulated hypertrophy [82].
An acute bout of RE induces the proliferation of the satellite cell pool already within 24 h after stimulation [83], which significantly accumulates SC content when repeating RE for 1 month [84]. The SC pool then remains stable after pausing RE for up to 30 days, indicating an RE–induced preconditioning of skeletal muscle for up to a month after RE–induced priming of skeletal muscle [84]. SCs are essential for skeletal muscle adaptability, especially in older adults [75], and the capillary supply surrounding SCs is critical for their responsiveness to RE in aging muscle [85]. A combination of RE and high-intensity interval training on a cycle ergometer has shown to increase SC responsiveness in older adults by increasing capillary supply [75]. Importantly, RE alone can induce an increase in capillary density in muscle fibers in older subjects [86], and preserve capillary content, which was increased after previously conducted high-intensity ET [87].
Repeated RE can further change the myofiber distribution within skeletal muscle [88-90]. Human skeletal muscle comprises slow oxidative type I, fast oxidative-glycolytic IIa, and fast-glycolytic IIx myofibers [91]. Although the dominant protein that allows quick classification of muscle fibers is the myosin heavy chain (MyHC) [92], a multitude of distinct protein isoforms contribute to the composition of fast and slow myofibers [93].
An important characteristic of myofibers is their ability to gradually shift their phenotype by transitioning from or into a hybrid fiber state, comprising several MyHC isoforms [94]. The underlying mechanism involves a calcium-dependent signaling pathway that translates neuromuscular activity into changes in fiber-type-specific gene expression [95,96].
Repeated contractions lead to the elevation of sarcoplasmic levels of Ca2+ [97] within active muscle fibers, which activates calmodulin. This, in turn, activates calcineurin, a phosphatase that dephosphorylates the nuclear factor of activated T-cells (NFAT), enabling it to enter the nucleus and regulate the transcriptional activity of “slow genes” coding for slow fiber type I proteins [98]. Several isoforms of NFAT regulate different subsets of fiber type-specific genes in myofiber myonuclei [99]. Depending on the increased or decreased neuromuscular activity (e.g., by duration, intensity, fiber recruitment [100], or inactivity [101]), there is a permanently changing pattern in the expression of fast and slow genes within skeletal muscle fibers [98].
When a clear pattern of either frequently repeated muscle contractions or inactivity is sufficiently maintained over an extended period, accumulated gene expression alters the protein composition of those skeletal muscle fibers [102]. This eventually causes a detectable transition from one fiber type to another, hereby also altering their functional properties [103,104].
While endurance exercise predominantly transforms myofibers from fast to slow [105], 12–16 weeks of RE training induced both, fast to slow, from IIx to IIa [89], and also slowto fast-shifting, from I to IIa [89,90], in young and older human subjects.
It is generally accepted that RE increases the proportion of IIa fibers in human skeletal muscle [88,106]. It should be noted, though, that most RE–induced fiber transitions within this time course occurred by reducing the hybrid fibers and shifting them to a fiber type containing only one MyHC isoform [90].
Keypoints:
• RE is a highly potent stimulus that exerts effects on skeletal muscle tissue widely beyond muscle growth
• RE modulates the extracellular matrix and the sarcomeric cytoskeleton to adapt to mechanical strain
• RE modulates the satellite cell pool to maintain long-term growth and repair potential
• RE modulates fiber type composition, mitochondrial function and capillarization to adapt its metabolic capacity under recurring stimulation
Regulation of muscle mass by resistance exercise and dietary protein administration
Hypertrophy results from accumulated myofibrillar protein synthesis when RE is repeated for several weeks and when protein breakdown is exceeded [107].
Muscle protein synthesis is regulated by the molecular activation of translation in skeletal muscle [42], which is controlled by the mTOR signaling pathway [108]. The mTOR complex consists of two major components, mTORC1 and mTORC2, which are regulated by interacting proteins [109,110]. mTORC1 phosphorylates the downstream proteins 4E-BP1 and p70S6K, which act on further targets and contribute to the initiation of protein translation [111] (Figure 1 (8)). p70S6K phosphorylates ribosomal protein S6 (rpS6), which is a crucial component of the ribosome itself and initiates translation [112] (Figure 1 (7)).
The inhibition of the mTOR pathway by rapamycin blocks RE–induced protein synthesis in humans, highlighting its crucial role in skeletal muscle growth [113].
Within the mTOR pathway, multiple signals converge [114] (Figure 1 (8)) and are integrated to optimize protein synthesis depending on the cellular environment. The essential amino acid leucine can activate mTOR signaling [115], and contraction-induced signals increase leucine uptake via the LAT1 amino acid transporter after 12 weeks of RE [116] (Figure 1 (5)). However, ingesting essential amino acids in the absence of muscle contraction by RE does not induce significant muscle growth [117]; therefore, the training component remains essential. However, the administration of dietary protein significantly adds to the re-induced increases in protein synthesis, and thus, muscle anabolism [118-120]. Increased ingestion of dietary proteins (up to 1.5 grams per kg body weight) in combination with RE–based skeletal muscle stimulation is beneficial for muscle maintenance and growth, especially in aging populations [121]. Recent studies reported that single doses of up to 40 grams of protein ingested after RE may be beneficial [120]. This is particularly relevant for the aging population, which generally has lower sensitivity and potential to absorb proteins via digestion [122].
The growth factor IGF-1 was initially believed to dominantly activate mTOR and protein synthesis after RE via activation of protein kinase B (AKT) [123] (Figure 1 (4)). Indeed, transgenic models with artificial AKT activation induce significant hypertrophy [124]. In 2008, however, it was argued that the IGF-1 pathway may not be the only mechanism that induces muscle growth, as animals with unfunctional IGF-1 receptors still developed hypertrophy [125]. Although still under investigation, mechanical signals are nowadays accepted as important stimuli for muscle protein synthesis after RE [18].
In addition to the dominant growth-controlling mTOR pathway, the giant titin molecule also contributes to skeletal muscle adaptation in response to RE–induced mechanical tension [126] (Figure 1 (1 & 2)). Titin proteins contain a conserved kinase region that influences diverse signaling pathways. Among these, calcium-dependent degradation as well as proteasomal protein degradation pathways (e.g., MuRF1) are regulated [126], so that titin controls sarcomeric protein turnover to some degree. A recent study highlighted the mathematical simulation of the titin-dependent contribution to muscle growth [127].
Protein synthesis can also be activated independently of mTOR [128,129]. By circumventing mTOR, focal adhesion kinase (FAK), which is part of the force-sensing cytoskeleton of the sarcomere, can transduce mechanical signaling towards p70S6K activation [130], and MAP kinases can directly activate p90s6k, which phosphorylates rpS6 [131] (Figure 1 (6)). From the perspective of practical experience, the importance of mechanical stimulation for muscle growth is obvious because weight-bearing contractions are conducted in basically all training settings that aim to induce relevant muscle growth.
RE activates mTOR signaling within 15–30 min after an acute exercise session [132,133]. The magnitude of this response depends on the volume [134] and intensity of the RE [135] and is essential for activating protein synthesis. The phosphorylation of mTOR-related signaling proteins usually returns to baseline within 24–48 h in human muscle [132], but may be extended up to 48 h in rat muscle under leucine administration [136]. Within this timeframe, muscle protein synthesis is elevated up to 48 [137] or even 72 h in human skeletal muscle [58]. However, the magnitude of the acute mTOR-related signaling response declines rapidly when training is repeated for at least 3 weeks [138]. We determined that this reduction in signaling persists as long as RE is not paused for at least 10 days [138]. The molecular mechanisms underlying this phenomenon are unclear; however, we argue that the cytoskeletal adaptation of myofibers may dilute the mechanical strain in myofibers and thus reduces the mechanically induced activation of mTOR. These findings indicate that the timeframe of a microcycle without RE stimulation is sufficient to re-sensitize skeletal muscle signaling responses.
Individual muscle fiber responses accumulate signals in the global response, which can be detected in the entire skeletal muscle via western blotting [139]. Myofibers show a highly specific p70S6K- and rpS6-phosphorylation pattern after RE [132,140]. Intense and eccentric RE induces p70S6K phosphorylation predominantly in type II fibers [141], whereas submaximal RE augments signaling in type I fibers [132].
All myofiber types can increase in size upon repeated RE, but type II fibers generally exhibit a greater potential for muscle growth [142]. Conversely, aging- or inactivity-induced muscle fiber atrophy affects type II more than type I fibers [143,144]. The molecular reasons for the greater growth-potential of type II fibers are still under study, as these fibers have a lower protein synthesis capacity than type I fibers [145]. Recently, it has been argued that the higher glycolytic capacity of type II fibers may contribute to the greater hypertrophic potential via a redirected glycolytic flux [146,147], which increases the capacity to produce amino acids and nucleotides to support growth conditions via glycolytic intermediates [51].
Previous findings showed that eccentric RE effectively enhanced strength gains [148 and anabolic responses [132]. However, myofibrillar damage would likely increase when performing eccentric resistance training and plyometric or power-associated exercises [149,150].
Although intense RE and especially eccentric contractions strongly induce mTOR signaling and protein synthesis, mechanical strain constantly threatens myofibrillar integrity [14]. Significant damage can occur within the muscle fibers, depending on the state of adaptation as well RE intensity [33,34,37] (Figure 2).

Skeletal muscle tissue of the human vastus lateralis muscle in the rested state (left) and after intense resistance exercise (right). (A) Electron microscopic picture of skeletal muscle in the rested state showing regular z-disk alignment in undamaged sarcomeres. (B) Electron microscopic picture of human skeletal muscle 24 h after intense RE, exhibiting myofibrillar damage, z-disk streaming, and irregular orientation of fibrils. (C) Fluorescence staining of the z-disk marker FLNC (green) of a human skeletal muscle fiber at rest in the untrained and undamaged state. (D) Fluorescence staining of the z-disk marker FLNC (green) and Xin-repeat binding protein (XIRP1) (red) as a marker of myofibrillar damage in a myofiber 60 min post intense RE. Note that the overlay (orange) clearly shows the abundance of a myofibrillar lesion in which XIRP co-localizes within a strongly damaged area of sarcomeres.
Keypoints:
• RE–induced muscle growth is predominantly regulated by the activation of mTOR-signaling, which increases protein synthesis for up to 72 h
• Mechanical stimulation of the skeletal muscles can increase protein synthesis also independently of mTOR and can induce the growth of type I and II fibers
• The administration of protein via means of nutrition enhances mTOR signaling and protein synthesis
• mTOR signaling responses are significantly reduced when RE is repeated for several weeks
Resistance exercise threatens skeletal muscle integrity
Skeletal muscle damage can occur in various degrees. RE–induced myofibrillar damage cannot be easily detected or quantified using simple methods. Early studies on skeletal muscle damage in young subjects using biopsy samples were published in 1976 [151]. Skeletal muscle damage induced by exercise is commonly accompanied by a later onset (12– 24 h after RE) of pain sensations in muscles [36], known as delayed-onset muscle soreness (DOMS). DOMS is usually associated with increased creatine kinase (CK) levels, peaking at 24–72 h post-RE [152]. Therefore, CK levels are generally considered an indirect marker of muscle damage [152].
However, CK levels may not precisely reflect the degree of muscle damage, as they are subject to considerable inter-individual differences [153,154]. CK levels also increase after endurance exercise [155,156]; notably, potentially without inducing any DOMS symptoms []157,158. Skeletal muscle damage is further associated with intramuscular swelling and extended immune responses [41] that are not mechanistically associated with CK release. Therefore, the role of CK as a reliable marker of damage assessment remains questionable [159].
In the RE community, muscular damage associated with DOMS has long been assumed to be a requirement to induce hypertrophy, following the “no pain, no gain” paradigm (as discussed, e.g., by Flann et al.) [160]. To date, no clear relationship has been detected between muscle damage and hypertrophy, and most data show that muscle damage induced by RE is not a prerequisite for enhancing the hypertrophic potential [160,161]. Furthermore, the underlying causes of DOMS remain controversial and are likely multifactorial [36,162]. It was recognized early that DOMS symptoms are rapidly reduced when RE of the skeletal muscle with the same or similar intensity is applied. This phenomenon, referred to as the “repeated bout effect” [37], can be observed after unfamiliar exercise, e.g., downhill running [163], but also after defined RE sessions [164-166].
At the structural level, muscle damage manifests as a derangement of z-disks (Figure 2 (B)) and disruption of filaments; observations were made as early as 1976 [151,167]. At the molecular level, the derangement of sarcomeres and z-disks is associated with the partial or complete unfolding of proteins within the sarcomeric network [72,168].
Myofibrillar damage can manifest as the abundance of small and highly localized lesions, areas in which most of the infrastructure is damaged, and specific proteins accumulate [169,170]. Among these, Xin actin-binding repeat-containing proteins 1 and 2 (XIRP1 and 2) bind to unfolded proteins and stabilize these clients, similar to a transient cellular glue. The Xin proteins are excellent damage markers that can be used to quantify lesions in muscle [169,171]. XIN-proteins are also located at satellite cells and support the activity of their proliferation [172].
This acute response of XIRP is essential to initiate the recruitment of important proteostatic proteins, e.g., heat shock protein B5 (HSPB5), to sites of damage (own observations) (Figure 2 (D)), from which these chaperones also control the initiation of CASA [173].
Muscle damage induces a rapid response of macrophages, which infiltrate the defective areas and activate a highly coordinated immune response [41]. This response leads to the initiation of macrophage-dependent degradation of defective proteins [41] and cytokine-stimulated activation of SCs to proliferate, differentiate, and re-establish muscle regeneration [174]. The mechanisms of muscle regeneration and adaptation are tightly linked and orchestrate a highly controlled molecular regulation that is initiated within minutes or hours after an acute bout of RE [48,175].
Keypoints:
• RE can induce muscle damage, which is associated with the delayed onset of muscle soreness (DOMS)
• DOMS is associated with a coordinated immune response that induces cytokine-mediated activation of macrophages and satellite cells to initiate muscle repair
• Repeated RE helps skeletal muscles to adapt to the imposed stress and reduces DOMS symptoms
• Specific proteins, such as chaperones and Xin-repeat binding proteins (XIRP), localize to sites of damage and initiate mechanisms to maintain and re-establish skeletal muscle tissue homeostasis
Molecular pathways that coordinate the early regulation of proteostasis after an acute bout of resistance exercise
The activation of specific signaling pathways after RE also expands to the activation of mechanisms regulating protein degradation [176]. The cytoskeletal arrangement of sarcomeres is highly complex and prone to contraction-induced mechanical damage; therefore, a machinery that controls damaged proteins is required. In general, skeletal muscle proteins undergo high protein turnover, which is further enhanced by RE [177,178]. Protein degradation in skeletal muscle depends on three pathways: the ubiquitin -proteasomal pathway (UPP) [177], the calcium-mediated calpain pathway [179], and autophagy [180]. The UPP plays a central role in muscle protein breakdown (MPB) and atrophy [181]. Within this system, the E3-ubiquitin ligases muscle RING-finger 1 (MuRF1) and muscle associated F-box atrogin-1 (MAFbx1), which are localized at the m-band and z-disks of the sarcomere, are essential components of myofibrillar breakdown (Figure 1 (11)).
There is a high degree of interaction between proteolytic pathways and calpain-mediated protein breakdown, e.g., it is assumed to prime sarcomeric proteins for further degradation via UPP [107]. Hence, along with a rapid increase in protein synthesis after RE, MPB too, is rapidly activated in skeletal muscle [107]. While MPB is activated to a greater extent in untrained than in trained subjects [182], breakdown rates are reduced within 24 h after RE, whereas protein synthesis extends for up to 48 h [107]. Hence, a higher synthetic rather than degradation response generally explains the higher net protein balance observed after RE [107]. Therefore, the acute aftermath of RE is characterized by the contemporaneous regulation of protein synthesis and breakdown, also known as protein turnover.
Another mechanism that is rapidly activated in acutely stressed tissues is the unfolded protein response (UPR) [40]. The UPR is activated via exercise-specific signals, starvation and high-fat diets [40,183]. It is an unspecific and generalized stress response that links the disruption of the endoplasmic reticulum (ER) membrane (sarcoplasmic reticulum [SR] in muscle) with the activation of gene expression and protein synthesis. Three ER transmembrane sensors, protein kinase R-like ER protein kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) [183] control the UPR. Under normal physiological conditions, these proteins remain inactive by binding to BiP/glucose-regulating protein 78 (GRP78) [184]. In response to ER stress, BiP dissociates and binds to misfolded proteins, thus enabling PERK, IRE1 and ATF6 to be activated. The UPR in humans is induced within 48 h after RE, but is reduced after repeated training [185], which is indicative of exercise-induced adaptation of the ER/SR after repeated stimulation. Although the UPR is critically important in restoring protein homeostasis, its specific role in enhancing skeletal muscle adaptation to RE has not yet been described.
Mechanical strain induced by RE can partially or fully unfold specific proteins and filaments within the sarcomeres, initiating controlled molecular events that target proteostasis restoration [43].
As mentioned previously, RE–induced mechanical strain can produce lesions that appear as spatially and clearly defined areas of damage distributed in skeletal muscle fibers (Figure 2 (D)). Within such lesions, Xin proteins, as well as small heat shock proteins and chaperones, such as CRYAB (HSPB5) and HSPB1 (HSP27), rapidly accumulate (own data, not yet published) after an acute bout of RE. Xin proteins can regulate SCs and myoblast function [172], but bind specifically to sarcomeric proteins such as actin and filamin [172,186]. Xin proteins further cross-link with F-actin, making it more resistant to depolymerization. Due to their dynamic redistribution in skeletal muscle in response to damage and their ability to regulate myoblast function, Xin proteins play an important role in skeletal muscle remodeling and repair.
As a first line of defense against the substantial degradation of damaged tissue, binding of small heat shock proteins, Xin proteins or co-chaperones to unfolded and damaged proteins prevents their aggregation and degradation [187]. Chaperones can furthermore refold proteins [188] or direct them to degradation via autophagy [189].
HSPB5 shows high sensitivity to mechanically induced stress, and its phosphorylation rapidly increases after a single bout of RE [190]. Phosphorylation of HSPB5 is mainly mediated by p38 MAP kinase (p38) [187], which increases the subcellular mobility of HSPB5 by disassembling HSPB5 oligomers into the monomeric state [191] (Figure 3).
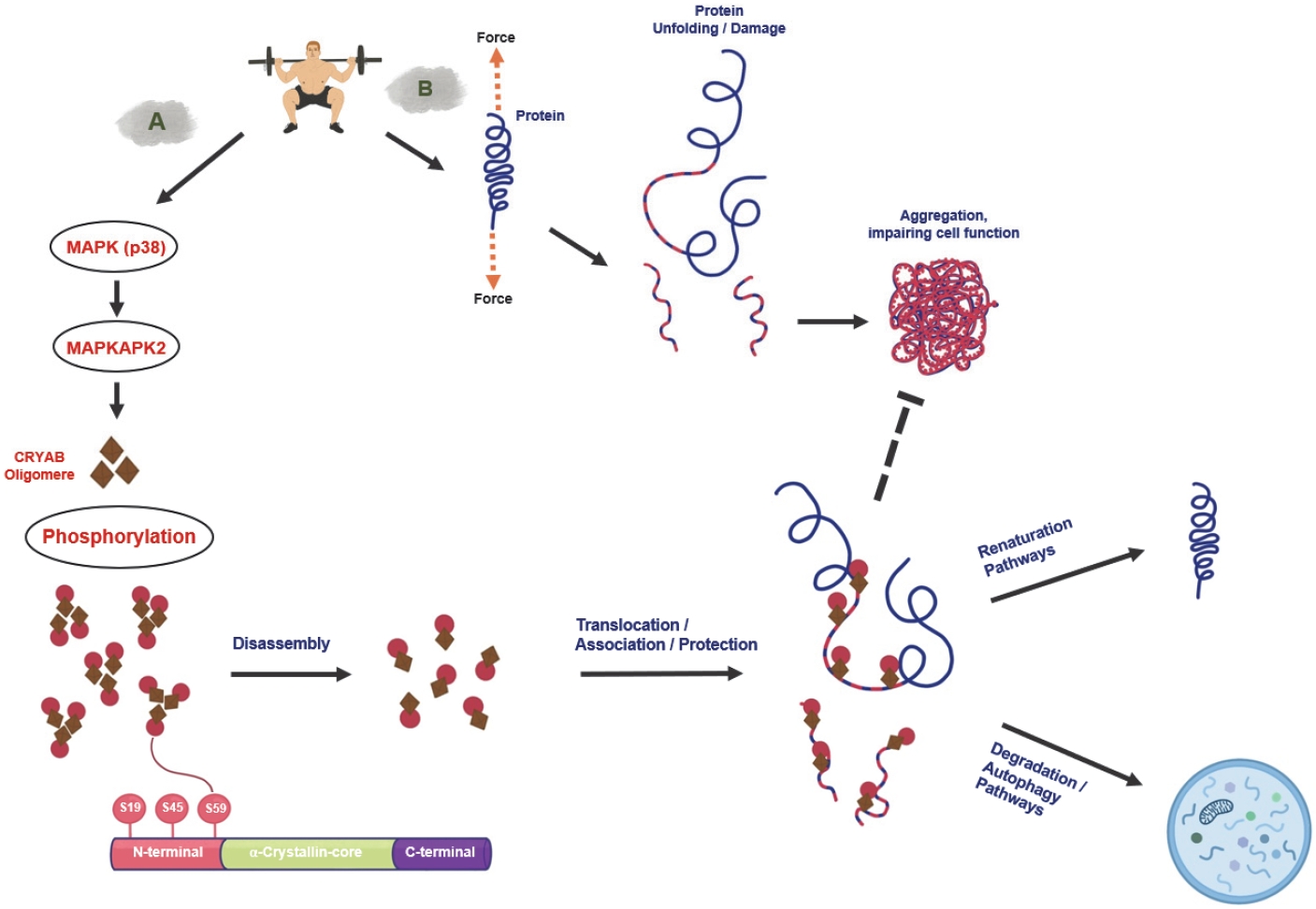
Resistance exercise activates small heat shock proteins, which control the renaturation of unfolded/damaged proteins and the degradation of terminally dysfunctional proteins via autophagic pathways. (A) RE stimulates p38 (p38 MAP-kinase)-phosphorylation, which activates MAPK-activated protein kinase 2 (MAPKAPK2). MAPKAPK2 phosphorylates the small heat-shock protein-beta 5 (HSPB5) at serine 59. HSPB5 is localized at the sarcomeric z-disk in the oligomeric state. Phosphorylation of HSPB5 can occur at multiple sites; however, phosphorylation at serine 59 leads to the disassembly of HSPB5-oligomers, bringing them into a more mobile state, from which they can translocate and associate to unfolded or damaged proteins. HSPB5 supports then the renaturation pathways or degradation of client proteins via interaction with other heat shock proteins. (B) RE mediates a force-induced unfolding of client proteins in skeletal muscle, e.g., titin, desmin and vimentin, leading to damage. In this condition, unfolded proteins can aggregate and impair cell function or be rapidly degraded. pHSPB5Ser59 phosphorylation prevents this, thereby supporting sarcomeric integrity.
We conducted several RE–based studies and determined that the phosphorylation of HSPB5 significantly increased as early as 15–45 min after RE [70,190]. This response occurs within different types of muscle fibers and is dependent on the intensity and volume of the RE [190]. While maximal and eccentric RE affects rather type II fibers, strength endurance training (commonly considered 15 or more repetitions [192]) affects type I fibers more [70,193]. The accumulated response in muscle generally increases with higher training volume [190]. Although discussed, we assume that the fiber-type-specific response also indicates the recruitment of fibers during RE and the mechanical stress these fibers have been exposed to during RE [190].
In a washout experiment using radioimmunoprecipitation assay (RIPA) buffer, we determined that HSPB5 is strongly bound to its clients in myofibers, especially after an unfamiliar bout of RE [70]. Under such conditions, the buffer did not remove HSPB5 from the myofibers. In contrast, repeated RE adapts the myofibrillar environment in a way that weakens the binding state, and RIPA treatment reduces HSPB5-binding [70]. This indicates that the phosphorylation and association of HSPB5 with client proteins are two distinct mechanisms that regulate acute responses to RE in muscle.
Within the sarcomeric cytoskeleton, the homodimer FLNC, which binds to actin in the z-disc, unfolds during RE–induced mechanical strain [72] (Figure 4). This exposes a specific binding motif for the co-chaperone BAG3, the WW domain [43]. BAG3 binding to FLNC recruits a collection of other co-regulatory factors, including SQSTM1 and HSPB8 [173], to form a protein complex, which then orchestrates FLNC degradation (Figure 4). This so-called CASA complex initiates the formation of a phagophore that engulfs the FLNC–BAG3 complex to prepare for its autophagosomal degradation [173]. We determined that intense eccentric RE, but not low-intensity RE, significantly degraded FLNC and BAG3 within 24 h after three sets of eight eccentric contractions [12]. This highlights the crucial difference between distinct RE intensities at the molecular level. BAG3 initiates FLNC degradation and also increases its expression via the activation of YAP and TAZ transcription factors [194] (Figure 4). We detected elevated FLNC RNA levels in the skeletal muscles of young subjects 24 h after RE [12].

Chaperone-assisted selective autophagy (CASA) is an essential mechanism to control, restore and maintain skeletal muscle proteostasis under conditions of recurring damage. Resistance exercise unfolds FLNC proteins in the sarcomere, which are localized in the z-disk to anchor actin filaments and interact with other proteins. The co-chaperone BAG3 (BAG family molecular chaperone regulator 3) is also localized at the z-disk and recognizes unfolded FLNC via its WW-binding domain. BAG3 binding to FLNC initiates its association with other proteins, thus forming the CASA complex. The CASA complex initiates phagophore and autophagosome formation and its subsequent lysosomal degradation. BAG3 further interacts with transcription factors that translocate into the nucleus to regulate the increased expression of FLNC, which is a prerequisite for FLNC substitution and its increased incorporation after repeated stimulation.
Importantly, in several studies, the acute molecular responses of mTOR signaling [138] and HSPB5 phosphorylation [70] were reduced throughout repeated RE.
These observations indicate the role of time-dependent molecular and structural skeletal muscle adaptations that feed back to the individual acute response of myofibers.
Keypoints:
• RE stimulates muscle protein turnover by initiating muscle protein synthesis and breakdown
• RE acutely stimulates the unfolded protein response (UPR) to link tissue damage with acute cellular responses of gene expression, protein synthesis, and protein stabilization
• RE is associated with acute increases in proteasomal protein degradation
• RE mediates rapid HSPB5 phosphorylation within type I and II skeletal muscle fibers to prevent the aggregation and degradation of damaged and unfolded proteins
• Acute resistance exercise-induced mechanical strain unfolds Filamin C in skeletal muscle z-discs, which activates CASA to degrade, substitute and incorporate FLNC at sites of sarcomeric damage
• Myofibrillar damage is a stimulus for adapting the sarcomeric cytoskeleton
Resistance exercise unfolds FLNC proteins in the sarcomere, which are localized in the z-disk to anchor actin filaments and interact with other proteins. The co-chaperone BAG3 (BAG family molecular chaperone regulator 3) is also localized at the z-disk and recognizes unfolded FLNC via its WW-binding domain. BAG3 binding to FLNC initiates its association with other proteins, thus forming the CASA complex. The CASA complex initiates phagophore and autophagosome formation and its subsequent lysosomal degradation. BAG3 further interacts with transcription factors that translocate into the nucleus to regulate the increased expression of FLNC, which is a prerequisite for FLNC substitution and its increased incorporation after repeated stimulation.
Repeated resistance exercise adapts the sarcomeric environment and alters the molecular regulation of muscle damage and adaptation
Interestingly, a sequence of only seven subsequent RE sessions induced a significant reduction in HSPB5-phosphorylation [70], along with reduced p70S6K and rpS6 phosphorylation [138], when human subjects underwent RE 3 times per week with a training intensity of approximately 80% of 1RM. This signaling response was reduced up to the 13th RE session and increased again only after a break of 10 days without RE. An explanation for this finding could be increased cytoskeletal stability of the sarcomere, decreasing mechanically induced signaling. Indeed, in previous studies, we detected increased FLNC [12] and desmin levels [70] after 7–10 repeated RE sessions (i.e., 2–3 weeks). The abundance of these proteins began to decline already after the aforementioned short timeframe without RE, indicating that the constitution of the sarcomeric cytoskeleton changed quickly in response to reduced loading.
This study reported increased myofibrillar damage after the first three RE sessions; however, it was reduced at the seventh RE session [12]. As we introduced above, the BAG3-mediated CASA machinery has, besides the acute task of degrading defective FLNC, also long-term effects on skeletal muscle, which increase FLNC protein levels after repeated RE. Along with increased desmin levels [70], incorporating these proteins is believed to enhance sarcomeric stability [39], likely reducing mechanical stress on the sarcomere and decreasing HSPB5-phosphorylation. Such mechanisms very likely contribute to the repeated bout effect and highlight the necessity of continued RE training to ensure the cytoskeletal adaptation of myofibers and reduced damage.
Skeletal muscle damage was assumed to be an important cellular trigger that stimulates muscle growth. However, recent data indicate that muscle damage is not essential in augmenting muscle hypertrophy [195]. In a 12-week RE–study, Damas et al. determined that substantial muscle growth was induced only after the reduction of initially occurring muscle damage and despite a time-dependent reduction in protein synthesis [44].
As muscle damage occurs more often in a fatigued state [35,196], the avoidance of substantial fatigue during RE might be vital for maintaining force development and reducing damage. The velocity-based approaches that are used to control RE loads yet provide important knowledge. It has been documented that RE which is conducted until muscular fatigue does not elicit more pronounced increases in muscle strength and growth than a condition “close to fatigue” [22,23,29,31]. A practical strategy to control this issue is described as “repetitions (reps) in reserve” [197,198], meaning that each set of RE is conducted until a point at which one to three more contractions will still be executable until complete muscle failure would be achieved. However, reaching that point exerts a sufficiently strong stimulation to ensure muscle adaptation.
As mentioned in previous chapters, the protein degradation machinery is rapidly activated after acute RE, implying that an adapted skeletal muscle would also reduce proteasomal degradation after repeated RE. Indeed, MPB is reduced when humans adapt to repeated RE [182]. Correspondingly, Stefanetti et al. observed reduced MuRF protein levels under adapting conditions in human skeletal muscle [199].
Keypoints:
• RE adapts the sarcomeric environment via FLNC and desmin incorporation, which is associated with a reduction in molecular signaling despite unchanged training intensity
• Myofibrillar damage is not considered a necessary stimulus to induce hypertrophy
• RE initially requires a reduction in myofibrillar damage before the onset of maximized net protein accumulation
• A short time frame without RE de-adapts the sarcomeric environment and re-sensitizes molecular signaling and chaperone responses in skeletal muscle
Changes in the molecular regulation of muscle growth after repeated resistance exercise
In the long term, augmented muscle growth is associated with the well-documented addition of new myonuclei via satellite cells [81], providing sufficient DNA for transcription. However, whether the addition of new myonuclei is indispensable for hypertrophy remains controversial, and recent analyses point to the rejection of this assumption [200,201]. Relevant findings in this context are derived from animal studies in which the artificial inhibition of the SC pool can be conducted [200].
In the history of this discussion, the myonuclear domain theory was introduced [202,203], which proposes that growing muscle will expand the volume of the muscle, so that the myonuclear domain (MND) may not provide enough mRNA for that given volume. In this case, the existing number of myonuclei would be insufficient to supply muscle fibers with mRNA to support protein synthesis in a hypertrophic muscle fiber. In contrast, more myonuclei would reduce the MND and support further growth. This theory is still under discussion, and it is unclear whether the addition of myonuclei is mechanistically and indispensably regulated by increased myonuclear domain size due to previous muscle growth.
Regardless of these issues, there is a well-documented observation of a rapid re-increase in strength and muscle mass after muscle loss when the muscle was previously strength-trained [204]. The search for the mechanisms behind that phenomenon, also known as “muscle memory,” is still ongoing, but two major aspects were identified contributing to a molecular explanation. In a pioneering mouse experiment, acquired myonuclei persisted for several weeks in skeletal muscles, even after immobilization-induced muscle loss [205]. This mechanism might very well contribute to the effect in skeletal muscle because DNA inherits the biological basis for storing information. Meanwhile, accumulated data indicate that myonuclei persist in human skeletal muscle even after weeks of detraining, and recent studies have highlighted that myonuclei do not undergo substantial degradation, and once they are incorporated, they persist [205-207].
In addition, epigenetic mechanisms have been shown to play a prominent role in preserving RE–induced DNA modifications over months following detraining. Epigenetic mechanisms modify the accessibility for transcription factors to the DNA [208]. Hypomethylation of CpG-rich islands increases accessibility, whereas hypermethylation decreases it [209,210].
A recent study by Seaborne et al. [209] reported that 7 weeks of RE, conducted three times per week, hypomethylated clusters of genes belonging to the mTOR pathway and the proteasomal degradation machinery. The resumption of RE resulted in an even bigger gain in muscle mass after 7 weeks of re-training when detraining-induced muscle loss was preceded by previous strength training. While acute resistance training-induced hypomethylation of >9000 CpG sites in the untrained state, >18,000 sites showed hypomethylation after RE with previous detraining [209].
Regardless of the scientific inconsistencies concerning the molecular mechanisms of muscle memory, a mixture of myonuclei-induced and hypomethylation-induced muscle memory might be responsible for the fact that prolonged periods of muscle training in humans may precondition skeletal muscle at the molecular level and support us in periods of reduced activity, e.g., during disease [211].
Muscle growth depends critically on the machinery that orchestrates protein synthesis. Ribosomes are activated to initiate translation under growth-stimulating conditions and through the mTOR pathway [212]. Although it has been shown that protein synthesis and mTOR-related signaling decline after acute bouts of RE when strength training is repeated [138,213], the capacity for protein synthesis also depends on ribosomal content [212]. Recent studies have shown that increased ribosome biogenesis accounts for a long-term increase in the translational capacity and efficiency of skeletal muscle to support growth [214] also under resting conditions [215]. This finding contributes to explaining why muscle growth occurs in later phases of repeated resistance training when the initial damage is reduced and increased translational capacity adds to increasingly blunted anabolic signaling and reduced protein synthesis [216]. Thus, the role of protein synthesis has also been considered to be controlled beyond the acute RE–induced increases in protein synthesis and breakdown.
Keypoints:
• RE–induced satellite cell proliferation and differentiation increases myonuclear content in skeletal muscle
• Skeletal muscle that was primed by repeated RE for several weeks inherits the potential for extended skeletal muscle memory, primarily mediated by epigenetic modifications
• Repeated RE increases ribosomal content, expanding the capacity for protein synthesis
Concluding remarks and practical considerations
Resistance exercise is a potent stimulus that can adapt skeletal muscle in the long term in terms of strength, muscle mass and energy metabolism. In order to control these functions, a variety of molecular regulators are coordinately activated in skeletal muscle, which are initially triggered by specific stimuli due to muscle contraction.
In the case of RE, particularly mechanical stimuli are exerted, which are crucial for inducing specific adaptations of skeletal muscle that cannot be induced by ET. Importantly, when we go to the gym, intense resistance training damages sarcomeric substructures significantly, requiring specific molecular regulators to maintain and re-establish tissue homeostasis. Such systems play essential roles in skeletal muscle adaptation and allow us to train again only a few days later under the conditions of a repaired muscle, which becomes increasingly primed towards repeated stimulation and may eventually lead to recognizable muscle growth. The growth develops only when the structural damage, which occurs after unaccustomed RE, is progressively reduced. From a global perspective, this may be obvious, considering that it is not useful to tear down the structures which are aimed to be built. Since training intensity and fatigue are critical components contributing to muscle damage, their application must be balanced, especially in view of the status quo of the skeletal muscle condition. In this context, some data suggest that a high training intensity during RE exerted until muscle fatigue is not always superior in terms of induction of muscle growth compared to lower intensities or when RE is carried out only close to fatigue.
Moreover, it is important to consider that when RE– based stimulation is repeated under equal conditions for approximately 3 weeks, the adapted sarcomere will exhibit reduced mTOR-related signaling, but also reduced muscle damage. The stimulation then hits a muscle in which various substructures have adapted, including ribosomal capacity, SCs, mitochondria, the sarcomeric cytoskeleton, and epigenetic modifications within myonuclei. Such adaptations may contribute to the extended muscle growth when mTOR-related signaling is already reduced. The orchestration of these adaptations shifts the structural basis of skeletal muscle to a trained state with increased strength, muscle mass and energetic performance, positively affecting general health.
Nevertheless, muscle biology is unrelenting and rapidly initiates deadaptation of sarcomeric substructures within 10 days, when RE is paused after a previous training program. Although muscle mass per se might not be reduced within this timeframe, it affects at least the sarcomeric cytoskeleton, making the sarcomere increasingly prone again to myofibrillar damage. This can be easily comprehended by anyone who has experienced muscle soreness after a session of RE preceded by an extended period without resistance training.
In recent decades, molecular exercise physiology and muscle biology have generated a huge amount of data that have enabled us to understand the time frame, function and diversity of mechanisms and substructures that adapt skeletal muscle tissue to RE. We now know that RE intensity, proximity to fatigue, training continuity and periods without training are variables that dynamically change skeletal muscle adaptation. These variables should be considered when creating RE programs for people of different age groups and training statuses. Small things can make a significant difference, also in resistance training.